Protein là nhân tố thiết yếu cho sự sống được hình thành từ các amino axit, sau đó trải qua quá trình gấp xoắn để hình thành cấu trúc 3D phức tạp. Chức năng của protein phụ thuộc chủ yếu vào cấu trúc 3D độc đáo của protein. Tìm hiểu về cấu trúc 3D của protein chính là một thách thức lớn trong 50 năm qua của các nhà khoa học. Trong một bước tiến mới gần đây, DeepMind - công ty trực thuộc Alphabet Inc. đã công bố một hệ thống AI mới với tên gọi Alpha Fold 2, có khả năng vượt trội trong việc giải quyết những thách thức lớn về tiên đoán cấu trúc protein.
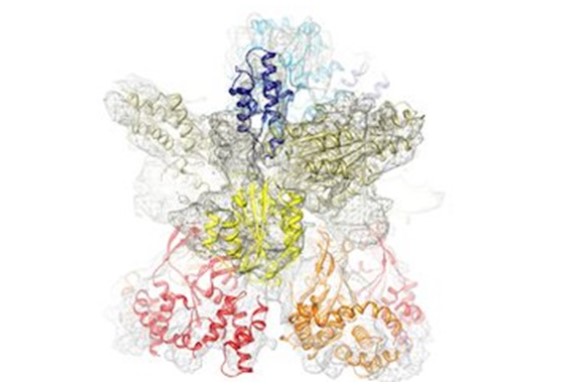
Hình 1. Phức hợp hai tiểu đơn vị của enzym magie chelatase. Cấu trúc này được xác định bằng phương pháp kính hiển vi điện tử lạnh (Cryo Electron Microscopy).
Điểm mấu chốt trong chức năng của protein là gì?
Trong cơ thể người và hầu hết các sinh vật hiện hữu, protein được cấu thành từ một chuỗi 20 loại amino axit cơ bản, là đơn vị cấu thành của nhiều cấu trúc, enzym và xúc tác cho nhiều phản ứng hóa học trong cơ thể sinh vật.
Sau khi trải qua quá trình dịch mã sẽ hình thành chuỗi amino axit là cấu trúc căn bản nhất trong quá trình tổng hợp protein. Sau đó, chuỗi amino axit này sẽ tiếp tục hoàn thiện cấu trúc bậc hai của protein bao gồm xoắn alpha và gấp beta. Các xoắn alpha và gấp beta kết hợp tạo thành cấu trúc bậc ba của protein, bước tiếp theo, hai hoặc nhiều chuỗi polypeptit (tiểu đơn vị polypeptit) sẽ hợp lại thành cấu trúc bậc bốn (hay còn gọi là cấu trúc oligo - oligomeric structure). Một oligo có thể bao gồm hai tiểu đơn vị giống nhau (homo-oligomer) hay khác nhau (hetero-oligomer).
Trình tự protein quy định phần lớn cấu trúc không gian ba chiều của protein. Tuy nhiên các biến thể khác nhau trong trình tự này vẫn có thể tạo ra các cấu trúc không gian ba chiều tương tự nhau, quy định các chức năng gần giống nhau. Xác định cấu trúc không gian ba chiều của protein có thể tiết lộ chức năng của protein đó, hơn nữa còn có thể dựa vào các mô hình tương đồng để dự đoán đáng tin cậy về cấu trúc của một protein mới.
Tại sao cấu trúc 3D của protein lại quan trọng đến vậy?
Cấu trúc 3D của protein chính là đặc điểm độc nhất vô nhị: không có bất kỳ protein nào giống với protein nào. Điều đó có nghĩa là mỗi trình tự chuỗi amino axit sẽ có cấu trúc 3D riêng biệt, đồng thời cấu trúc 3D của protein cũng xác định chức năng của protein. Khi protein có cấu trúc 3D bất thường nghĩa là chức năng của protein bất thường, và có nguy cơ gây bệnh. Cấu trúc 3D của protein quan trọng đến độ trong bài phát biểu của nhà hóa học người Mỹ Christian Anfinsen, khi ông nhận giải Nobel vào năm 1972 đã nhấn mạnh rằng, theo lý thuyết, trình tự amino axit của protein phải xác định đầy đủ cấu trúc của chính nó. Phát biểu này đã khơi mào cho một giả thuyết kéo dài suốt 5 thập kỷ qua về việc làm sao có thể tính toán cấu trúc 3D của protein chỉ dựa trên trình tự amino axit 1D của chính nó. Vấn đề ở đây là, theo nghịch lý Levinthal (ra đời vào năm 1969), một protein điển hình có tới 10300 cấu hình và hiện tại chúng ta có khoảng 200 triệu protein, trong số đó có 170.000 protein đã biết rõ cấu trúc 3D.
Để khảo sát cấu trúc 3D, các nhà nghiên cứu đã sử dụng nhiều phương pháp khác nhau, trong số đó nổi bật là cộng hưởng từ hạt nhân (nuclear magnetic resonance) và tinh thể học tia X (X-ray crystallography). Những kỹ thuật này, cũng như một số kỹ thuật mới ngày nay như kính hiển vi điện tử lạnh (cryo-electron microscopy) phụ thuộc rất nhiều vào quá trình thử - sai, có thể kéo dài nhiều năm và tiêu tốn hàng triệu USD. Ví dụ điển hình như để xác định cấu trúc 1 protein, phương pháp tinh thể học tia X tiêu tốn 120.000 USD và mất khoảng 1 năm.
DeepMind và Alpha Fold 2
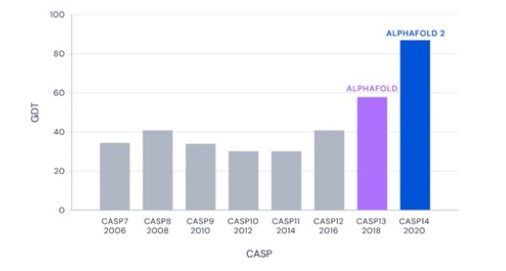
Alpha Fold 2 của DeepMind đã giải quyết được vấn đề cốt lõi trong cấu trúc 3D của protein kéo dài suốt gần 5 thập kỷ qua. CASP (Critical Assessment of Techniques for Protein Structure Prediction) là một cuộc thi nhằm đánh giá độ chính xác của các phương pháp tiên đoán cấu trúc 3D của protein. Độ chính xác của phương pháp được tính bằng giá trị khoảng cách tổng phân tử (Global Distance Test - GDT). Nói một cách dễ hiểu, GDT là tỷ lệ phần trăm khoảng cách ước tính của các amino axit so với vị trí chính xác của nó. Các protein được dùng làm tiêu chuẩn trong cuộc thi đều là những protein mới được xác định gần đây và chưa hề được công bố cấu trúc trên toàn thế giới. Trong kết quả đánh giá CASP lần thứ 14, Alpha Fold đã đạt số điểm 92,4 GDT trên tổng số các bài kiểm tra, riêng đối với bài kiểm cho các protein dạng khó, mang tính thách thức nhất, Alpha Fold đã đạt được điểm trung bình là 87 GDT.
Hình 2. Quá trình cải thiện độ chính xác trung bình trong tiên đoán cấu trúc 3D của Alpha Fold.
Thành tựu này có ý nghĩa sâu sắc, bởi đây là một bước tiến vĩ đại trong sinh học cấu trúc, đồng thời cũng là thành tựu lớn nhất của AI trong suốt hơn 20 năm qua. Thậm chí nhiều chuyên gia còn cho rằng, Alpha Fold 2 có thể chạm tới giải Nobel đầu tiên cho chuyên ngành học máy (machine learning).
Alpha Fold 2 có điểm gì khác biệt?
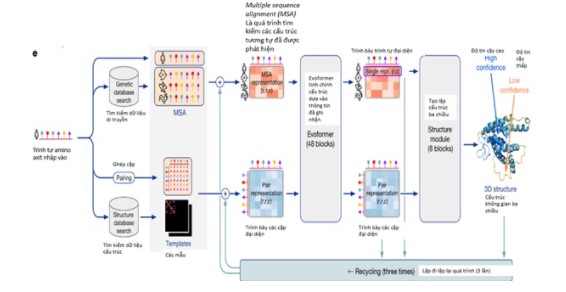
DeepMind đề cập một số điểm khác biệt căn bản của phiên bản Alpha Fold 2 mới nhất, vượt trội hơn so với phiên bản cũ, bao gồm một hệ thống mới có tên gọi attention-based neural network system (tạm dịch: hệ thống mạng neuron dựa trên chủ ý), quá trình thử nghiệm từ đầu đến cuối nhằm cố gắng đưa ra giả thiết về các biểu đồ tiềm ẩn. Alpha Fold 2 sử dụng các trình tự có liên quan về mặt tiến hoá, liên kết nhiều trình tự (multiple sequence alignment - MSA) và tái trình bày các cặp amino axit nhằm đưa ra một biểu đồ chuẩn xác nhất.
Hình 3. Cấu trúc của mô hình Alpha Fold 2.
Alpha Fold trực tiếp tiên đoán tọa độ 3D của tất cả các nguyên tử nặng cho một protein nhất định, bằng cách sử dụng trình tự amino axit chính và các chuỗi protein tương đồng làm dữ liệu đầu vào.
Hệ thống mạng lưới Alpha Fold gồm hai phần chính. Đầu tiên, phần trục của hệ thống xử lý các tín hiệu đầu vào thông qua Evoformer (là một khối cấu trúc mạng nơron mới gồm các lớp lặp đi lặp lại), tạo thành mảng Nseq x Nres (Nseq là số chuỗi; Nres là số phần tử dư lượng). Biểu diễn MSA được khởi tạo từ MSA thô. Evoformer bao gồm một số lượng các thành phần dựa trên chủ ý và không chủ ý (attention-based and non-attention based components), đưa ra giả thiết về các cấu trúc protein cụ thể sớm và liên tục hoàn thiện.
Tiếp theo phần trục của hệ thống là phần mô-đun cấu trúc để tạo lập cấu trúc 3D dưới dạng quay vòng và tiến hành dịch mã cho từng phần còn lại của protein. Những đổi mới chính trong phần mạng lưới này cho phép tinh chỉnh cục cùng lúc các phần của cấu trúc, phân tích về các phân tử chuỗi bên không được thể hiện và định hướng của các phần tử dư lượng.
Việc cải tiến lặp đi lặp lại toàn bộ mạng lưới, góp phần đáng kể vào cải thiện độ chính xác dù chỉ thêm một thời gian ngắn.
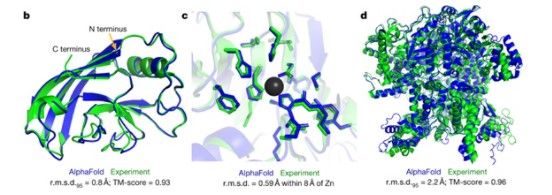
Hình 4. Tiên đoán về cấu trúc các protein trong CASP14. Màu xanh lam biểu hiện cho chuỗi protein tiên đoán bởi Alpha Fold 2, so với cấu trúc thực biểu thị bằng màu xanh lá. Các protein lần lượt: B.T1049 (PDB 6Y4F), C.T1056 (PDB 6YJ1), D.T1044 (PDB 6VR4).
Ứng dụng của Alpha Fold
Một số ứng dụng hữu ích của Alpha Fold trong thời điểm hiện tại có thể bao gồm tiên đoán một số cấu trúc protein của virut SARS-CoV-2, bao gồm protein ORF3a và gần đây nhất là ORF8. Hai cấu trúc protein này đã được so sánh với cấu trúc xác định bằng thực nghiệm và cho kết quả tin cậy cao.
Trong tương lai gần, hiểu biết về chức năng của protein 3D giúp đào sâu vào các chức năng chưa rõ của gen mã hóa protein đó. Đồng thời tìm ra nguyên nhân gây bệnh do sai sót trong cấu trúc gấp xoắn của protein. Alpha Fold 2 đưa ra phương pháp mới giúp thiết kế một protein nhanh chóng thay thế chức năng của protein lỗi, từ đó ứng dụng trong y học điều trị, nông nghiệp (protein diệt côn trùng, tạo lớp phủ bảo vệ thực vật khỏi sương giá), tái tạo mô (qua protein tự lắp ráp), chất bổ sung (cho sức khỏe và chống lão hóa) hoặc vật liệu sinh học.
Trong tương lai xa hơn, Alpha Fold 2 có thể được phát triển để tiên đoán mối tương tác giữa các protein và quá trình hình thành phức hợp protein, mô phỏng vật lý một cách chính xác các hệ thống sinh học (ví dụ như mô phỏng tế bào, cơ quan), vén màn các bí ẩn trong môi trường sinh học và nhân tạo.
Trần Thụy Hương Quỳnh
Đại học Y khoa Kansai, Osaka, Nhật Bản
TÀI LIỆU THAM KHẢO
1. https://deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology.
2. https://www.deepmind.com/blog/article/AlphaFold-Using-AI-for-scientific-discovery.
3. https://pubmed.ncbi.nlm.nih.gov/20223218/.
4. https://www.nature.com/articles/s41586-021-03819-2.